
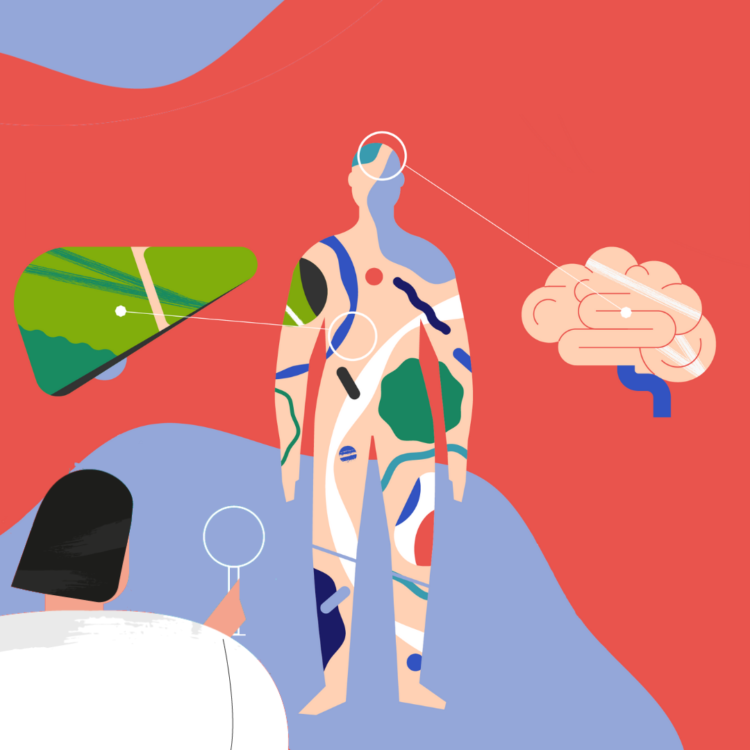
Doença de Wilson, inimiga do fígado e do cérebro
A doença de Wilson é uma doença rara, genética e hereditária que impede a eliminação do cobre na bílis conduzindo à sua acumulação nociva no organismo, sobretudo no fígado e no cérebro. Inicia-se no nascimento, com progressão gradual da doença, sendo a idade média de diagnóstico entre os 12 e os 23 anos. Estima-se que atinja um em cada 30.000 habitantes em toda a Europa.
Trata-se de uma doença hepática com sintomas neurológicos e psiquiátricos. Os sintomas relacionados com a doença hepática manifestam-se, em média, entre os 9 e os 13 anos e caracterizam-se por dor abdominal, icterícia (coloração amarela da pele e mucosas), ascite, edemas, hemorragia digestiva e alteração do estado mental.
Já os sintomas neuropsiquiátricos surgem em média entre os 15 e os 21 anos e estão relacionados com a dificuldade na articulação das palavras ou na coordenação motora, com a rigidez e tremor, escorrência involuntária da saliva, alteração da personalidade e mesmo depressão ou psicose.
São sintomas gerais da doença, hepáticos e/ou neuropsiquiátricos, a alteração das provas hepáticas, anemia hemolítica, níveis diminuídos de ceruloplasmina (transportador do cobre) no sangue, níveis elevados de cobre na urina de 24 horas e a existência dos chamados Anéis de Kayser-Fleischer (anéis castanhos ou verdes na córnea).
Se não for tratada, a Doença de Wilson pode ser fatal. Existe tratamento eficaz, que deverá ser contínuo, por toda a vida, mesmo na ausência de sintomas, e que passa pela toma de fármacos que aumentem a eliminação do cobre (penicilamina ou trientina) ou que reduzam a absorção intestinal do cobre (zinco).




